The FDA 483 Isn’t the Risk. The Response Is. Mar 2, 2026

Srilekha Deka, MD, PhD
Script Molecular is a biotech consulting firm in San Francisco, CA.

A step-by-step guide to understanding, responding to, and resolving FDA inspection observations before they escalate.
An FDA Form 483 is issued at the conclusion of a facility inspection when an investigator observes conditions that may constitute violations of the Federal Food, Drug, and Cosmetic Act (FD&C Act). It is not a final enforcement action but it is a serious regulatory signal that corrective action is both expected and overdue.
The Form 483 does not stand alone. FDA evaluates it alongside the Establishment Inspection Report (EIR), evidence gathered during the inspection, and the company’s written response.¹ How you respond and how quickly you respond, can determine whether the matter closes at this stage or escalates into something far more consequential.
How does the full process unfold?
.
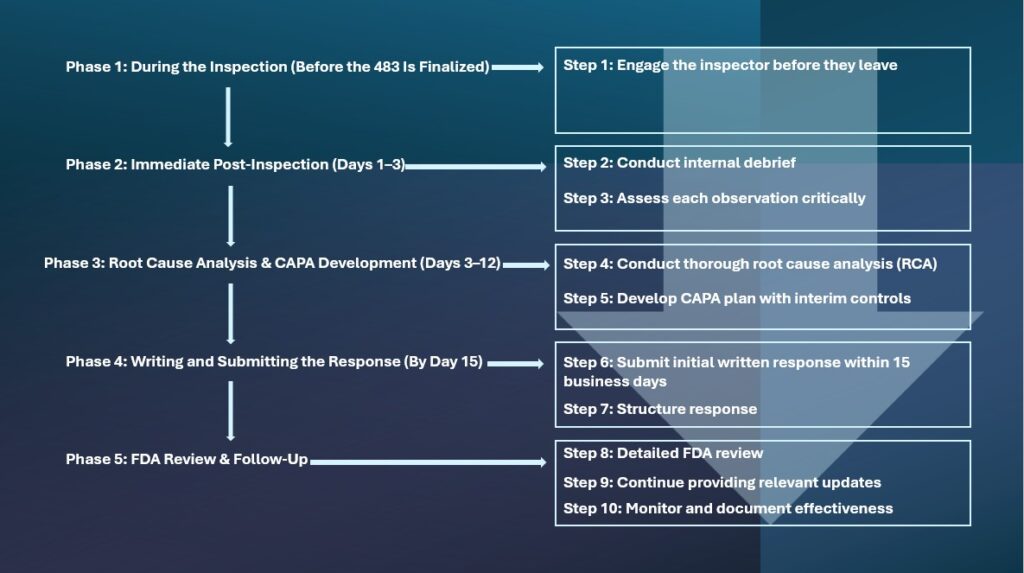
.
Phase 1: During the inspection (Before the 483 is finalized)
Step 1 — Engage before the inspector leaves
The exit meeting is critical and often underutilized. Before the investigator finalizes Form 483 it is important to:
- Clarify each observation and ask questions if the scope or intent is unclear.
- Ensure senior leadership understands the nature and severity of findings.
- Correct any factual misunderstandings on the spot, with documentation available.
Misreading an observation at this stage creates downstream problems. Alignment early, even if it is partial, can sharpen the company’s response and reduce the risk of escalation.²
.
Phase 2: Immediate Post-Inspection (Days 1–3)
Step 2 — Convene an Inspection Response Team (IRT)
Within 24 to 72 hours of inspection it is important to:
- Debrief with quality, regulatory, legal, and operational leadership.
- Capture all verbal comments made during the inspection, not just what appears on the 483.
- Review each observation with relevant subject matter experts.
- Identify documentation gaps, systemic exposures, and any related observations not cited.
The goal is not merely to draft a response. It is to understand what the inspection revealed about the state of the company’s quality system.
Step 3 — Assess each observation honestly
Before writing a single word of the response:
- Determine whether each observation is accurate.
- If accurate: define what remediation is required and at what urgency.
- If inaccurate: identify the objective evidence that supports the company’s position.
Evidence and transparency are what the FDA looks for.³
.
Phase 3: Root Cause Analysis and CAPA Development (Days 3–12)
Step 4 — Conduct a thorough Root Cause Analysis
Superficial fixes are among the most common and costly mistakes companies make in 483 responses. For each observation:
- Define the problem statement precisely.
- Document the full scope of the investigation.
- Apply structured tools such as 5 Whys, fishbone (Ishikawa) diagrams, or fault tree analysis.
- Identify the true systemic root cause, not just the proximate trigger.
This should be a thorough process and should not be rushed to meet the 15-day deadline if it means compromising analytical depth. A weak root cause analysis increases the likelihood of escalation.⁴
Step 5 — Develop a CAPA Plan with Interim Controls
Permanent corrective actions often take time. The company’s response must demonstrate that risk is being controlled. It is important to include:
- Immediate containment actions that have already been implemented.
- Interim controls that will be implemented like enhanced monitoring, additional testing, process holds, or heightened oversight.
- Permanent corrective and preventive actions with defined owners and timelines.
- Measurable effectiveness checks to verify that actions taken have achieved their intended outcome.⁵
The FDA looks for objective evidence that your processes are currently under control, along with a clear, structured plan to ensure sustained implementation, ongoing monitoring, and long-term compliance.
.
Phase 4: Writing and submitting the response (By Day 15)
Step 6 — Submit response within 15 business days
The FDA expects a written response within 15 business days of inspection. If the response is received within this window, the FDA will conduct a detailed review prior to any decision to issue a Warning Letter.⁶
If the observations are complex and require additional investigation, it is appropriate to request more time, but this must be communicated proactively and the company has to demonstrate that the investigation has been initiated.
Step 7 — Structure the response for clarity and completeness
A well-structured 483 response should:
- Address each observation individually and in order.
- State clearly whether the company agrees or disagrees with the observation, and why.
- Present corrective and preventive actions with realistic, committed timelines.
- Include evidence of actions already completed like revised SOPs, training records, audit data, environmental monitoring results.
- Describe how effectiveness will be measured and verified.
- Reflect visible leadership commitment, not just quality department language.⁷
Many response letters include a structured appendix that maps each observation to its root cause, CAPA actions, responsible owner, target completion date, and update cadence. This level of organization signals competence and seriousness to the agency.
.
Phase 5: The FDA Review and ongoing follow-through
Step 8 — The FDA evaluates the submitted response
If the response is provided in a timely fashion, the FDA will assess whether:
- Root causes are adequately identified and plausible.
- Corrective actions are comprehensive and proportionate to the risk.
- Timelines are reasonable and commitments are credible.
- Risk to product quality and patient safety is being controlled in the interim.
A strong, well-documented response can halt escalation at this stage. A weak or dismissive one often accelerates it.⁸
Step 9 — Follow through on every commitment
The initial response does not need to close every open item but every commitment that was made must be honored. Missed timelines, incomplete CAPAs, and broken follow-up schedules are among the most reliable predictors of Warning Letter issuance. The FDA notices and will respond to these flags.
Step 10 — Monitor, document, and stay inspection-ready
Repeat observations are one of the strongest triggers for Warning Letters and escalated enforcement.⁹ After the response is submitted:
- Monitor quality systems for related issues or recurrence.
- Maintain a complete compliance package: investigation reports, root cause documentation, CAPA records, effectiveness check results, and objective evidence.
- Treat the next inspection as if it begins the day after this one closes.
.
What Happens If the Response Falls Short?
If the FDA determines your response is inadequate, a Warning Letter is likely to follow. Warning Letters are public and visible to investors, customers, partners, regulators in other jurisdictions, and the press. The downstream consequences can include:
- Delayed or blocked product approvals (NDAs, ANDAs, 510(k)s)
- Import alerts restricting product entry into the U.S. market
- Consent decrees and injunctive actions
- Damage to export certificates and international regulatory standing
- Heightened scrutiny on all future FDA submissions and interactions¹⁰
In regulated industries, a company’s relationship with the FDA is an asset. Poorly managed 483 erodes it.
.
The Bottom Line
A Form 483 is not a crisis but it is a stress test of a company’s quality culture, it’s leadership’s commitment to compliance, and the organization’s ability to identify and correct its own failures.
Companies that respond with transparency, thorough root cause analysis, substantive corrective actions, and genuine accountability tend to resolve observations without escalation. Companies that treat the response as a paperwork exercise tend to find out what comes next.
In regulated industries, compliance is not just about passing inspections. It is about demonstrating to the agency, to the market, and to the patients that the organization is in control.
.
REFERENCES:
- 1. FDA — Establishment Inspection Reports (EIR) and the 483 Process
https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/fda-form-483-frequently-asked-questions - 2. NSF International — Ten Steps to an Effective FDA 483 Response
https://www.nsf.org/knowledge-library/ten-steps-effective-medical-devices-fda-483-responses - 3. Gardner Law — Best Practices for Responding to FDA Form 483
https://gardner.law/news/best-practices-for-responding-to-fda-form-483s - 4. Greenlight Guru — How to Respond to FDA Form 483 in 7 Steps
https://www.greenlight.guru/blog/fda-483-inspection-observations-response - 5. FDA Guidance for Industry: Investigating Out-of-Specification (OOS) Test Results for Pharmaceutical Production
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/investigating-out-specification-oos-test-results-pharmaceutical-production-level-2-revision - 6. Inspection Observations
https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/inspection-observations - 7. FDA — Regulatory Procedures Manual: Warning Letters
https://www.fda.gov/media/71878/download - 8. Goodwin Law — Form FDA 483 Response Best Practices (June 2024 Draft Guidance Analysis)
https://www.goodwinlaw.com/en/insights/blogs/2024/06/form-fda-483-response-best-practices-announced-by-the-fda - 9. The FDA Group — FDA Officials’ Tips on Responding to 483s (2024 MedCon)
https://insider.thefdagroup.com/p/fda-officials-offer-tips-on-responding - 10. FDA — Import Alert Overview
https://www.accessdata.fda.gov/cms_ia/ialist.html